
.png)



If you’re developing software for healthcare, then it’s critical to know whether your product qualifies as Software as a Medical Device (SaMD) because once it does, you’re no longer just building an app. You’re entering a space where regulatory compliance, safety standards, and clinical validation become non-negotiable.
SaMD plays a direct role in diagnosing conditions, recommending treatments, or managing diseases. That kind of influence comes with serious responsibility and a complex set of global rules that must be followed.
Understanding how regulators define SaMD, what documentation you’ll need, and how to prepare for approval can save you time, money, and setbacks later. It also ensures your product meets the highest safety and quality standards, something every patient deserves.
This guide walks you through the entire SaMD development process—from classification and risk assessment to clinical evaluation and post-market surveillance—so your software stands up to scrutiny and succeeds in the healthcare market.
Key Takeaways
- Software as a Medical Device is standalone software that performs medical functions without being part of hardware medical devices, requiring strict regulatory compliance.
- Different regions have specific requirements for SaMD, with the FDA in the US, MDR in Europe, and PMDA in Japan each having distinct classification systems and approval processes.
- SaMD is categorized based on risk level and intended use, directly impacting the regulatory pathway and compliance requirements necessary for market approval.
- Creating compliant SaMD requires specialized expertise in both healthcare regulations and software development. Pi Tech's specless engineering approach can simplify this complex process.
- The global SaMD market is projected to reach $5.0 billion by 2033, creating significant opportunities for healthcare technology companies with the right development partner.
What is Software as a Medical Device (SaMD)?
Software as a Medical Device, or SaMD, refers to software that’s intended for medical purposes, without being part of a physical medical device. It can run on a smartphone, computer, or cloud platform and still be considered a medical device in its own right.
The key factor is your software’s intended use. When it’s designed to diagnose conditions, monitor vital signs, guide treatment decisions, or help manage chronic illnesses, regulators see it as a medical device, and that means stricter oversight.
This definition comes from the International Medical Device Regulators Forum (IMDRF), which makes it clear: software that only controls hardware (like an insulin pump or MRI machine) isn’t SaMD. But software that independently performs medical functions? That qualifies.
Understanding this distinction early helps you avoid costly mistakes during development.
The demand for SaMD is rising fast. According to a 2024 report, the global Software as a Medical Device market was valued at $1.4 billion in 2023, and it's projected to reach $5.0 billion by 2033, growing at a CAGR of 13.6%. That growth reflects how essential software has become in healthcare, from remote diagnostics to AI-powered clinical decision tools.
What is Considered a Medical Software?
Just because software is used in healthcare doesn’t automatically make it a medical device. To be considered medical software, your product must serve a specific medical purpose, and that purpose needs to be clear in how you label, describe, or market it.
Here’s how regulators typically define it:
- Your software has a declared medical purpose, such as diagnosing, preventing, monitoring, or treating a condition.
- It goes beyond basic data storage or communication—it analyzes, interprets, or generates patient-specific outputs.
- It has the potential to influence clinical decisions, directly impacting how a condition is diagnosed or managed.
- It produces individualized insights or outputs tied to a person’s health status.
On the other hand, software used for administrative tasks—such as appointment scheduling, billing, or storing general health content—isn’t treated as a medical device. It might be used in a healthcare setting, but it doesn’t perform a regulated medical function.
Knowing where your software fits is crucial. It determines your responsibilities and the kind of regulatory path you’ll need to follow.
How to Determine if Software is a Medical Device?
Before diving into development, it’s critical to confirm whether your software qualifies as a medical device. This shapes everything—from how you design it to the regulatory pathway you’ll follow.
Start with a clear, step-by-step review:
- Check the Intended Use Statement: Look at how the software is described in your documentation, marketing materials, and user instructions. Any mention of diagnosis, treatment, or patient monitoring could trigger medical device classification.
- Match Functionality to Regulatory Definitions: Compare what your software does against definitions provided by agencies like the FDA, EU MDR, or other regional authorities. Even small features can change how your product is classified.
- Assess the Level of Patient Risk: Ask yourself: what happens if the software fails or gives the wrong output? If there’s any chance it could lead to harm, even indirectly, regulators will likely treat it as a medical device.
- Analyze Its Role in Clinical Decision-Making: Is the software simply displaying data, or is it analyzing, guiding, or influencing decisions about patient care?
To make things easier, the FDA offers a Digital Health Policy Navigator, a free online tool that helps you assess whether your software falls under its regulatory scope. It’s a great starting point to avoid surprises later in development.
How Does Software Qualify as a Medical Device?
Knowing how your software is classified sets the stage for everything that follows. The key factor regulators look at is intended use—how you describe your software’s purpose and the functions it performs.
Your software likely qualifies as Software as a Medical Device (SaMD) when it meets these criteria:
- Serves a Medical Purpose: Designed to diagnose conditions, suggest treatments, monitor diseases, or support clinical decision-making.
- Acts Independently of Hardware: Performs its function without being part of or controlling physical medical equipment.
- Processes Patient-Specific Data: Analyzes, interprets, or generates outputs based on individual health data, providing insights that directly impact a patient’s care.
On the other hand, software that only stores, transfers, or archives data, like basic Electronic Health Record (EHR) systems, usually doesn’t qualify as SaMD. The same goes for tools built purely for administrative use, such as scheduling or billing platforms.
Getting this classification right ensures you follow the appropriate regulatory path—and helps you avoid setbacks later in development.
What is a Software-Defined Medical Device?
A software-defined medical device—sometimes called a software-only medical device—is essentially another term SaMD. What sets it apart is that all of its medical functionality is defined by software alone, with no need for dedicated hardware to operate.
In other words, the “device” is the software.
Here’s what defines it:
- Runs on Standard Hardware: Can operate on general-purpose platforms like smartphones, tablets, or cloud servers—no custom-built hardware required.
- Performs Medical Functions Through Code: Every diagnostic, analytical, or clinical function is handled entirely by software algorithms.
- Connects to Other Inputs: May receive data from external sources like sensors, wearables, or medical imaging tools, but it doesn’t depend on them to qualify as a medical device.
This approach makes the device easier to deploy, update, and scale, but it also means the software must meet the same safety and regulatory standards as traditional medical hardware.
SaMD vs. Software in a Medical Device (SiMD)
It’s easy to confuse Software as a Medical Device (SaMD) with Software in a Medical Device (SiMD)—but they’re not the same, and the difference matters when planning your regulatory strategy.
Here’s how they compare:
Software as a Medical Device (SaMD):
- Works Independently: Functions on its own to perform medical tasks without being part of any specific hardware.
- Runs on Standard Platforms: Can operate on smartphones, tablets, desktops, or cloud servers.
- Regulated by Its Own Purpose: Evaluated based on what the software is intended to do for medical use.
- Examples Include: A mobile app that analyzes skin conditions, or a cloud-based tool that detects irregular heart rhythms from uploaded ECG data.
Software in a Medical Device (SiMD):
- Embedded in Hardware: Built into the physical device it controls, such as scanners, pumps, or surgical robots.
- Essential to Device Functionality: Without it, the hardware wouldn’t work or deliver its intended results.
- Regulated as Part of the Device: Assessed together with the hardware as a combined medical product.
- Examples Include: Firmware inside an MRI machine, or the software controlling an insulin pump.
Understanding this distinction early helps you choose the right development process, testing protocols, and regulatory documentation, from start to finish.
SaMD Regulatory Framework Around the World
Medical device regulations vary widely around the world, and Software as a Medical Device is no exception. While many high- and middle-income countries, including the United States, EU member states, the UK, China, Australia, and Brazil, have either established or are actively developing formal regulatory frameworks specific to SaMD, the global picture is far from uniform.
In low- and middle-income regions, regulatory systems for SaMD are often still in early stages. For example, according to WHO, only 18 out of 34 surveyed countries (about 53%) have a national law covering medical devices, including SaMD. Even fewer—just 9 countries (22.5%)—have all the recommended regulatory documents in place.
In many parts of the world, regulation is still guided by "soft-law" approaches, such as guidelines, position papers, and voluntary standards, rather than enforceable, formal rules. This can create challenges for developers aiming for international distribution, as expectations may shift dramatically from one country to another.
Understanding each region's approach is key to planning your market access strategy and ensuring compliance across jurisdictions.
Does SaMD Require FDA Approval?
In the United States, SaMD is regulated by the Food and Drug Administration (FDA), and it typically requires either approval or clearance depending on its risk classification.
Here's how the FDA classifies SaMD:
- Class I (Low Risk): Most are exempt from premarket review, but they must still follow general controls, such as labeling, manufacturing practices, and record-keeping.
- Class II (Moderate Risk): Requires 510(k) clearance, which is the most common regulatory pathway for SaMD. This involves demonstrating that your software is substantially equivalent to an existing legally marketed device.
- Class III (High Risk): Requires Premarket Approval (PMA)—a much more rigorous process involving clinical data and detailed safety evidence. Only a small number of SaMD products fall into this category.
Most SaMD products are classified as Class I or II:
- Many Class I SaMDs are exempt from premarket submission but must still comply with FDA's general controls.
- The 510(k) pathway is the standard route for moderate-risk SaMDs and accounts for the majority of regulated software.
- PMA is rare, reserved for high-risk devices with the potential to cause serious harm if they fail.
Knowing where your software fits in this structure is critical. It affects everything from design and documentation to market launch timelines and post-market obligations.
United States (FDA)
The FDA regulates SaMD under its broader medical device framework with some digital-specific enhancements:
FDA Risk Classification System:

The FDA has also established innovative programs through its "Digital Health Innovation Action Plan," including:
- Digital Health Software Precertification (Pre-Cert) Program: Focuses on evaluating the software developer rather than each individual product
- Software Precertification Pilot Program: Testing regulatory approaches that accommodate faster development cycles
- Clinical Decision Support Software Guidance: Clarifying which decision support tools require regulation
European Union (MDR)
In the European Union, Software as a Medical Device is regulated under the Medical Device Regulation (EU MDR). Compared to the previous directive, the MDR introduced much stricter requirements, especially for software products.
Under the MDR, SaMD is classified into four risk-based categories:
- Class I: Low-risk software (e.g., general wellness or simple health monitoring apps)
- Class IIa: Medium-low risk (e.g., fertility tracking apps that provide time-window predictions)
- Class IIb: Medium-high risk (e.g., software monitoring vital signs that could trigger intervention)
- Class III: High-risk (e.g., software that drives or influences critical treatment, such as a closed-loop insulin delivery system)
The MDR places particular emphasis on clinical safety and performance. Even lower-risk SaMD must now meet higher regulatory expectations than before.
Key changes affecting SaMD under MDR include:
- Stronger Clinical Evidence Requirements: You must provide clinical data to demonstrate the safety, performance, and benefit of your software, especially if it influences treatment decisions.
- Expanded Post-Market Surveillance: You're expected to actively monitor your device after launch and document findings in periodic safety update reports (PSURs).
- Unique Device Identification (UDI): SaMD products must now include UDIs to improve traceability across the EU market.
- More Detailed Documentation: This includes comprehensive technical files, risk management plans, and a clinical evaluation report (CER) that justifies your software’s intended use and performance.
Navigating MDR can be complex, but meeting these standards positions your product for safe, trusted access to the EU healthcare market.
Japan (PMDA)
In Japan, Software as a Medical Device is regulated by the Pharmaceuticals and Medical Devices Agency (PMDA), using a four-tier risk classification system similar to those in other major markets:
- Class I: General medical devices with minimal potential for harm
- Class II: Controlled medical devices with moderate risk
- Class III: Highly controlled medical devices with substantial risk
- Class IV: Highly controlled medical devices with potentially fatal risk
Your SaMD’s classification depends on how it impacts patient care, and the level of risk if the software fails or produces incorrect results. Class I typically includes non-critical wellness software, while Class IV is reserved for life-supporting or life-sustaining functions.
Japan is also known for its forward-thinking approach to regulating innovative technologies. One standout example is the Sakigake designation—a program designed to expedite the review of breakthrough medical products, including cutting-edge SaMD, that show exceptional promise in treating serious conditions.
To access Japan’s market, you’ll need to align your product with MHLW (Ministry of Health, Labour and Welfare) regulations, undergo PMDA review based on risk level, and prepare robust clinical documentation, especially for higher-risk classes.
International Medical Device Regulators Forum (IMDRF)
The International Medical Device Regulators Forum (IMDRF) plays a leading role in shaping how Software as a Medical Device is regulated worldwide. Its goal is simple but critical: create consistent, harmonized standards that help regulators and developers align across different countries.
The IMDRF framework is now widely adopted or referenced by authorities in the United States, European Union, Japan, Canada, Australia, and other key markets. At the core of this framework is a risk-based classification matrix based on two main factors:
- Significance of the Information: How much the SaMD influences clinical decisions (e.g., driving treatment vs. just informing diagnosis)
- State of the Condition: Whether the condition is non-serious, serious, or critical
By combining these two elements, the IMDRF defines four risk categories—from Class I (lowest risk) to Class IV (highest risk). These categories help regulators determine the appropriate level of scrutiny and documentation required during the approval process.
For developers, this framework offers a more predictable regulatory path—especially when launching in multiple countries. As IMDRF continues pushing for global harmonization, the hope is to reduce duplicate work, speed up market access, and ease compliance burdens across jurisdictions.
SaMD Classification and Risk Categories
Understanding how your software is classified by risk is one of the most important steps in determining its regulatory pathway. While exact requirements vary by region, many authorities now refer to the IMDRF classification framework as a global standard.
IMDRF Classification Framework
The International Medical Device Regulators Forum (IMDRF) classifies SaMD based on two core factors:
- Healthcare Situation or Condition: Is the condition non-serious, serious, or critical?
- Significance of the Information Provided: Does the software treat or diagnose, drive clinical management, or simply inform clinical management?
These two dimensions are combined in a matrix to define the SaMD risk category:

Your software’s position in this matrix determines how much regulatory oversight it will require, from premarket documentation to post-market monitoring.
SaMD Risk Categories Explained
Category I (Lowest Risk):
- Software that informs clinical management for non-serious conditions
- Example: Software that calculates body mass index (BMI) to inform general fitness goals
Category II:
- Software that drives clinical management for non-serious conditions or informs management for serious conditions
- Example: Software that analyzes heart rhythm data to detect potential arrhythmias for further evaluation
Category III:
- Software that treats or diagnoses non-serious conditions or drives management for critical conditions
- Example: Software that processes MRI images to detect and highlight potential tumors for radiologist review
Category IV (Highest Risk):
- Software that treats or diagnoses critical conditions
- Example: Software that autonomously analyzes cardiac rhythms and delivers defibrillation commands to an implanted device
The higher the risk category, the more stringent the regulatory requirements, including quality management systems, clinical validation, and post-market surveillance..
7 Real-World Examples of Software as a Medical Device
Understanding SaMD becomes clearer when examining real-world examples that have successfully navigated regulatory pathways. These applications demonstrate the diverse potential of software in healthcare while providing insights into implementation strategies
To better understand the scope and potential of SaMD, let's explore some notable examples across different medical specialties and risk categories:
1. Diagnostic Imaging Analysis Software
Advanced AI algorithms that analyze medical images like X-rays, MRIs, or CT scans to detect abnormalities or assist in diagnosis. These systems can identify potential issues that human reviewers might miss or provide decision support for radiologists.
- Example Product: Aidoc's AI-powered solution for detecting and prioritizing critical conditions in CT scans, which received FDA clearance and is used in hospitals worldwide.
- Regulatory Status: FDA 510(k) cleared
- Risk Classification: Class II (moderate risk)
2. Mobile Diagnostic Applications
Smartphone applications that utilize device sensors or user input to diagnose or screen for medical conditions.
Example Product: FibriCheck, an FDA-cleared and CE-marked mobile application that uses a smartphone's camera to detect atrial fibrillation and other heart rhythm disorders.
This category demonstrates the accessibility benefits of SaMD, with a 2022 Mayo Clinic study showing that AI-guided mobile ECG screening detected previously undiagnosed atrial fibrillation in 7.6% of high-risk adults over 65 during short-term monitoring.
3. Clinical Decision Support Software
Programs designed to assist healthcare providers in making clinical decisions by processing patient data against medical guidelines or using algorithms to generate patient-specific assessments.
- Example Product: Epic's Deterioration Index, which analyzes patient vital signs and lab results to predict and alert clinicians about patients at risk of deterioration.
- Impact: At the University of Pennsylvania Health System, implementation of advanced clinical monitoring and predictive tools has contributed to improved patient management and may help reduce unexpected ICU transfers, reflecting the potential of algorithm-based early warning systems to enhance critical care outcomes.
4. Remote Patient Monitoring Systems
Software platforms that collect, analyze, and interpret data from wearable sensors or patient-reported outcomes to monitor health conditions remotely.
Example Product: Dexcom G6 Continuous Glucose Monitoring System's software component, which processes data from glucose sensors to provide real-time glucose level information and trends.
According to the American Diabetes Association, continuous glucose monitoring (CGM) helps people with diabetes spend about 70% of their day-roughly 17 hours-in the target glucose range, which is significantly more time in range compared to standard monitoring methods.
5. Digital Therapeutics
Software interventions designed to treat medical or psychological conditions, often through behavioral modification or cognitive exercises.
- Example Product: Pear Therapeutics' reSET, the first FDA-authorized prescription digital therapeutic for substance use disorder.
- Clinical Evidence: In clinical trials, reSET demonstrated a 40.3% abstinence rate at the end of 12 weeks of treatment when combined with outpatient therapy, compared to a 17.6% abstinence rate for standard outpatient therapy alone in patients with substance use disorder (excluding primary opioid users).
6. Radiation Treatment Planning Software
Programs that help oncologists plan radiation therapy by calculating optimal radiation doses and treatment areas based on patient imaging.
Example Product: Varian's Eclipse Treatment Planning System, which helps clinicians design highly conformal radiation treatment plans for cancer patients.
7. Surgical Planning and Navigation Software
Software that processes patient images to create 3D models for surgical planning or provides real-time guidance during procedures.
Example Product: Brainlab's Surgical Navigation System, which provides surgeons with real-time guidance during neurosurgical procedures.
A 2025 retrospective study of 492 pedicle screws found that navigation systems reduced any screw deviation from 21% (non-navigation) to 8.4% (navigation), and deviations greater than 2 mm from 12% to 3.2%, representing roughly a 60–70% reduction in misplacement risk.
Each of these examples demonstrates the vast potential of SaMD to transform healthcare delivery while highlighting the importance of proper regulatory compliance based on their risk classification and intended use.
Key Regulatory Requirements for SaMD Development
Building Software as a Medical Device isn’t just about great code—it’s about creating software that meets strict regulatory expectations. To bring your SaMD to market and keep it there, you’ll need to follow a structured approach covering quality, risk, performance, and security.
Below are the key regulatory elements you’ll need to address during development:
1. Quality Management System (QMS)
A robust QMS conforming to standards like ISO 13485 is mandatory for SaMD development. This system must document processes for:
- Design and development controls
- Risk management
- Document control
- Supplier management
- Corrective and preventive actions (CAPA)
- Post-market surveillance
Your QMS should be tailored to software development, incorporating Agile or other methodologies, while maintaining regulatory compliance.
2. Risk Management
Comprehensive risk management following ISO 14971 is essential throughout the SaMD lifecycle. This involves:
- Identifying potential hazards
- Estimating and evaluating associated risks
- Implementing and verifying risk control measures
- Monitoring effectiveness of controls
For SaMD, special attention must be given to cybersecurity risks, data integrity issues, and algorithm validation.
3. Clinical Evaluation
Depending on the risk classification, clinical evidence may be required to demonstrate:
- Clinical validity
- Analytical performance
- Clinical performance
This might involve literature reviews, clinical investigations, or real-world performance studies. Higher-risk SaMD typically requires more substantial clinical evidence.
4. Software Development Life Cycle (SDLC)
SaMD development should follow a defined SDLC with appropriate documentation:
- Software requirements specification
- Software architecture and design
- Implementation and verification
- Integration testing
- System testing
- User acceptance testing
- Validation
The IEC 62304 standard provides a framework for software lifecycle processes for medical device software.
5. Cybersecurity Measures
As SaMD often processes sensitive patient data, robust cybersecurity is crucial:
- Threat modeling and security risk assessment
- Security requirements and design
- Secure coding practices
- Security testing
- Vulnerability management
- Incident response planning
FDA and other regulators increasingly scrutinize cybersecurity measures during the approval process.
6. Post-Market Surveillance
Once your SaMD is on the market, you must maintain:
- Complaint handling processes
- Vigilance reporting
- Periodic safety update reports
- Software update management
- Field safety corrective actions when needed
Effective post-market surveillance helps identify and address unforeseen risks while providing data for continuous improvement.
Common Challenges in SaMD Development
Developing Software as a Medical Device presents unique challenges that extend beyond typical software development. Understanding these challenges is crucial for planning effective development strategies:
Regulatory Complexity and Uncertainty
The regulatory landscape for SaMD continues to evolve as agencies adapt to rapid technological advances.
This creates several challenges:
- Interpreting regulations that may not explicitly address novel technologies
- Keeping pace with updated guidance documents
- Navigating different requirements across global markets
- Uncertainty about how regulations will be applied to innovative products
Many healthcare startups struggle with regulatory strategy because they lack expertise in interpreting and applying these complex frameworks to their specific software products.
Quality and Documentation Requirements
The documentation burden for medical devices is substantial compared to consumer software:
- Extensive traceability from requirements to testing
- Comprehensive risk management documentation
- Detailed design history files
- Software verification and validation records
- Clinical evidence documentation
These requirements can significantly extend development timelines and increase costs if not properly managed from the beginning.
Balancing Innovation with Compliance
One of the most significant challenges is maintaining innovation while satisfying regulatory requirements:
- Agile development practices must be adapted to incorporate regulatory controls
- Change management processes can slow down iterative development
- Validation requirements may limit the speed of feature deployment
- Risk management may constrain certain innovative approaches
Finding the right balance requires specialized expertise in both regulatory affairs and software development methodologies.
Algorithm Validation and Performance
For SaMD that utilizes artificial intelligence or machine learning:
- Demonstrating algorithm performance across diverse populations
- Validating algorithms against clinically relevant endpoints
- Addressing algorithm drift and continuous learning challenges
- Explaining algorithm decision-making to regulators and users
Regulatory bodies are still developing frameworks for evaluating AI/ML-based SaMD, adding complexity to the validation process.
Cybersecurity and Data Privacy
Given the sensitive nature of health data, SaMD faces heightened security challenges:
- Implementing robust security controls without compromising usability
- Meeting varying privacy regulations across different jurisdictions
- Managing security throughout the software supply chain
- Planning for security updates and vulnerability management
Failures in this area can lead to regulatory actions, legal liability, and loss of market trust.
How Pi Tech Helps Overcome SaMD Development Challenges
At Pi Tech, we understand the complex intersection of healthcare regulations and software development that makes SaMD projects particularly challenging. Our specialized approach addresses these challenges head-on:
Specialized Healthcare Regulatory Expertise
Our team brings deep expertise in healthcare compliance frameworks, including HIPAA, FDA regulations, and international standards like ISO 13485 and IEC 62304. This knowledge is crucial for SaMD development, where regulatory strategy must be integrated from day one.

Unlike traditional development partners that might treat regulations as an afterthought, our experience with healthcare compliance means we build regulatory considerations into the foundation of your software.
This proactive approach prevents costly rework and regulatory delays that often plague SaMD projects.
Senior-Only Development Teams
Pi Tech employs exclusively senior-level developers with extensive experience in regulated industries. This matters tremendously for SaMD projects where expertise in both technical implementation and regulatory requirements is essential.
How Our Senior Teams Make a Difference:
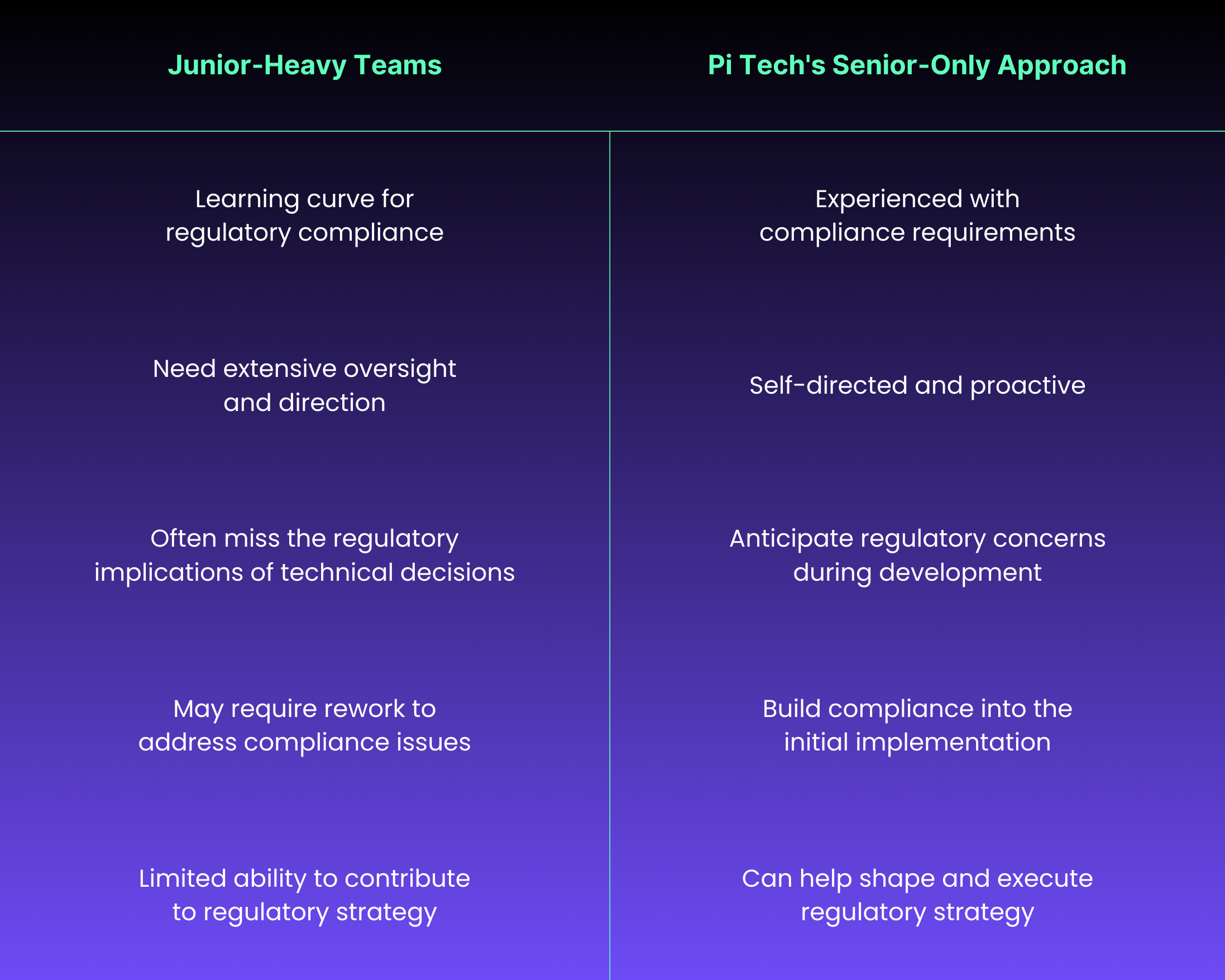
Our senior developers understand the critical importance of:
- Building traceability into the development process
- Implementing appropriate risk controls
- Creating maintainable, well-documented code that supports regulatory submissions
- Designing verification and validation protocols that satisfy regulatory expectations
This expertise translates to more efficient development cycles and higher-quality submissions when seeking regulatory clearance.
Innovative Specless Engineering Methodology
Our proprietary specless engineering methodology is particularly well-suited for SaMD development. Rather than requiring extensive documentation before writing a single line of code, we focus on understanding your objectives and collaboratively working toward solutions.
"Traditional development approaches often fail in healthcare because they can't adapt to the evolving regulatory landscape and clinical requirements. Pi Tech's specless engineering methodology gave us the flexibility we needed while maintaining compliance." — CTO of a digital health startup client
This approach allows us to:
- Start building solutions even with incomplete information
- Adapt quickly to evolving regulatory guidance
- Maintain compliance while demonstrating progress
- Balance innovation with regulatory requirements
For SaMD developers, this means faster time-to-market without compromising compliance—a critical advantage in the competitive healthcare technology landscape.
End-to-End Project Ownership
We don’t just code and walk away. Pi Tech takes full ownership of your SaMD project, from early planning through post-market support. You get a single partner accountable for strategy, execution, and compliance at every stage.
This comprehensive approach includes:
- Strategic Planning Phase
- Regulatory strategy development
- Quality management system implementation
- Risk management planning
- Development Phase
- Design and development with appropriate controls
- Iterative prototyping with regulatory considerations
- Implementation of required technical and quality controls
- Verification Phase
- Comprehensive verification and validation testing
- Technical documentation preparation
- Internal audits and gap analysis
- Regulatory Submission Phase
- Submission preparation and support
- Regulatory correspondence management
- Response to regulatory questions
- Post-Market Phase
- Post-market surveillance planning
- Complaint handling procedures
- Update and maintenance planning
This end-to-end capability eliminates the need to coordinate multiple vendors and ensures consistent compliance throughout the product lifecycle.
Developing Your SaMD Strategy: Next Steps
Creating a successful Software as a Medical Device requires a strategic approach that balances regulatory requirements with innovation and market needs. Here are the essential steps to develop an effective SaMD strategy:
1. Clarify Your Intended Use and Risk Classification
Before diving into development, clearly define your software's intended use and determine its likely risk classification:
Key Questions to Consider:
- What specific medical purpose will your software serve?
- Which patient populations will benefit?
- What clinical decisions will rely on your software?
- What would be the consequences of software failure or inaccuracy?
This foundational step will inform every subsequent decision in your development process. Higher-risk classifications require more rigorous controls and evidence, directly impacting timelines and budgets.
2. Develop a Comprehensive Regulatory Strategy
Based on your risk classification and target markets, create a regulatory roadmap:
Regulatory Strategy Checklist:
- [ ] Identify which regulatory pathways apply to your SaMD
- [ ] Determine the evidence requirements for each target market
- [ ] Establish timelines for regulatory submissions
- [ ] Budget for regulatory costs including submission fees and consulting
- [ ] Evaluate accelerated pathways like FDA Breakthrough Devices designation
A well-planned regulatory strategy prevents costly redirections later and ensures efficient use of development resources. The FDA's Digital Health Innovation Action Plan provides useful guidance for companies developing regulatory strategies for innovative digital health products.
3. Implement Quality Management Systems Early
Don't wait until development is complete to implement quality processes:
- Establish a right-sized QMS that complies with ISO 13485
- Develop SOPs for design control, risk management, and change control
- Create templates for required documentation
- Train your team on regulatory requirements and quality processes
- Consider electronic QMS to streamline compliance
Early QMS implementation ensures that you're collecting the necessary documentation throughout development rather than reconstructing it retroactively.
4. Choose the Right Development Partner
The right development partner can dramatically improve your chances of SaMD success:
Essential Partner Evaluation Criteria:

Pi Tech specializes in regulated healthcare software. Our senior-only model and proprietary specless engineering methodology allow us to move quickly, without compromising compliance. We embed regulatory thinking into every step, helping you reduce risk, shorten timelines, and launch with confidence.
5. Plan for Lifecycle Management
Launching your SaMD is only the beginning. Regulatory bodies, especially the FDA and the EU MDR, expect ongoing oversight of your software after it reaches the market. A well-designed lifecycle management plan helps protect patients, reduce compliance risk, and keep your product competitive.
Critical Post-Market Activities:
- Safety Monitoring
- Implement complaint handling procedures
- Establish adverse event reporting mechanisms
- Conduct periodic safety reviews
- Updates and Maintenance
- Define change control processes for software updates
- Develop validation protocols for patches and upgrades
- Determine when modifications require regulatory notification
- Security Management
- Create vulnerability monitoring procedures
- Develop incident response plans
- Implement regular security assessments
- Performance Monitoring
- Collect real-world performance data
- Analyze usage patterns and user feedback
- Track clinical outcomes where applicable
According to FDA guidance on Postmarket Management of Cybersecurity in Medical Devices, effective lifecycle management is not just a regulatory requirement but a critical component of patient safety.
6. Secure Necessary Resources
Even the best SaMD strategy can stall without the right resources behind it. Planning for budget, timeline, expertise, and infrastructure up front is essential to stay on track and avoid costly surprises.
Resource Planning Considerations:
- Financial: Budget for development, verification, validation, and regulatory submissions
- Timeline: Allocate sufficient time—SaMD development typically takes 18-36 months
- Expertise: Ensure access to clinical, regulatory, and technical specialists
- Infrastructure: Consider specialized testing environments and tools
- Partners: Identify needs for outside consulting or specialized services
According to recent industry data, around 70% of software projects exceed their initial budgets, with average overruns of 27%. Underestimating regulatory complexity and validation costs is one of the top reasons projects go off track, so plan accordingly.
Ready to Advance Your SaMD Project?
Developing Software as a Medical Device presents unique challenges that require specialized expertise in both healthcare regulations and software development.
At Pi Tech, we've helped numerous healthcare technology companies navigate these complexities and bring innovative SaMD products to market.
Our senior-only talent model ensures that your project is handled by experienced professionals who understand the critical intersection of software development and regulatory compliance. Our proprietary specless engineering methodology allows us to adapt quickly to evolving requirements while maintaining the documentation and controls needed for regulatory success.
Whether you're developing diagnostic software, a digital therapeutic, or a clinical decision support system, our team can guide you through the entire process—from initial concept to regulatory submission and beyond.
Ready to discuss your SaMD project? Contact Pi Tech today to learn how we can help you navigate the regulatory landscape and bring your healthcare software innovation to market.


